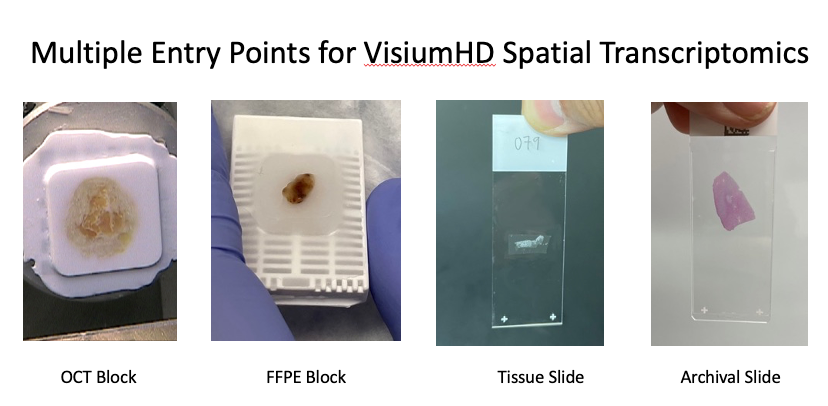
Submit your tissue samples and we’ll handle everything – from slide preparation and imaging to sequencing and spatial analysis. Receive high-resolution spatial gene expression maps with full data outputs and expert support.
Why work with 3DG for Spatial Transcriptomics?
Spatial transcriptomics projects depend on getting every step right. From tissue processing and high-quality imaging through library prep, sequencing, read mapping, and downstream analysis, a failure at any stage affects the whole dataset. We process FFPE and fresh frozen samples in-house from intake through histology and imaging, conduct the CytAssist probe hybridization and transfer process, construct the VisiumHD libraries, manage the sequencing and conduct the Space Ranger read mapping and clustering and additional follow-on analyses requested by the client. Every project dataset includes all raw sequence data, all Space Ranger output files, and additional informatics analysis – cell type assignment, spatial domain identification, and UMAP clustering and cell-centric analyses from segmented spatial data, ready for your team to use.
VisiumHD is 10x Genomics’ whole-transcriptome spatial platform, capturing gene expression at 2um bin resolution across fresh frozen or FFPE tissue sections. Whereas imaging-based platforms like Xenium interrogate targeted subsets of genes, VisiumHD measures every gene without requiring a predefined panel, making it the right choice for discovery work where you don’t yet know which genes matter.
3DG has run VisiumHD on a wide range of tissue types and sample preparations. As part of our analytical work, we have characterized segmentation noise in spatial transcriptomics data, quantifying the extent of transcript diffusion and comparing its effects across VisiumHD and imaging-based platforms. Our segmentation error whitepaper describes the findings in detail.
Preserves spatial location of transcripts within the tissue.
Deep and broad coverage – millions of transcripts captured across the full tissue section in a single assay.
Compatible with fresh-frozen and FFPE samples, expanding sample options. Available in 6.5mm and 11mm capture area formats.
Integrates imaging and sequencing for a more complete biological picture.
Enables discovery of novel gene expression relationships in cell niches, tissue organization, and disease microenvironments.
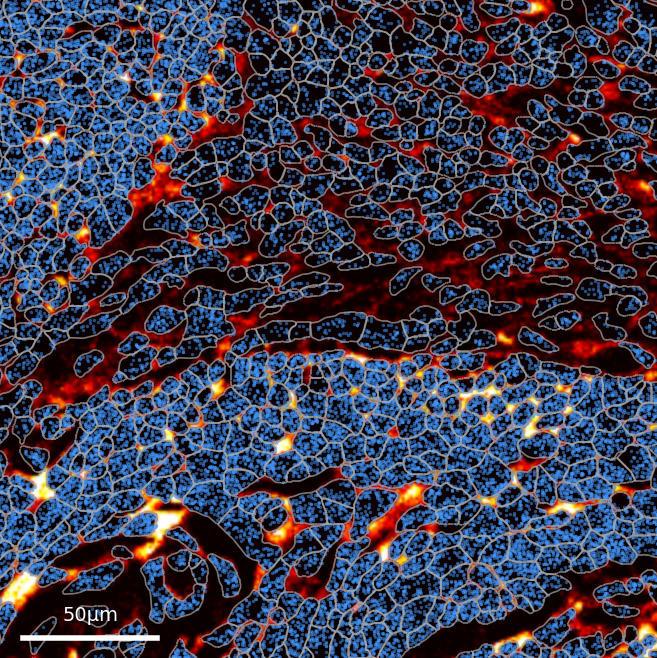
Cell-level segmentation enables the same analyses as single-cell RNAseq, UMAP clustering, differential expression, and cell type mapping, with spatial location preserved.
Beyond Human and Mouse
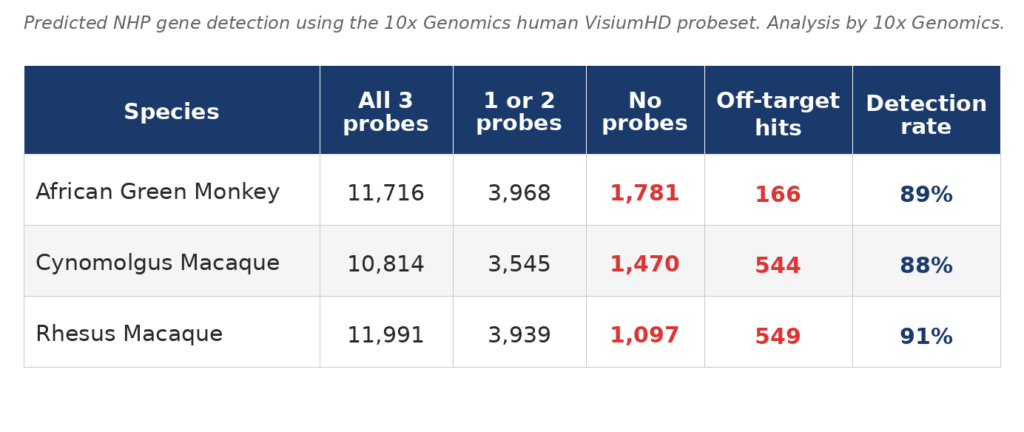
3DG has validated VisiumHD on non-human primate tissue using the human probeset. The table below shows predicted gene detection rates across three NHP species, based on probe homology analysis performed by 10X. Each gene is detected by 3 probes and about 90% of NHP genes are predicted to be detected by at least one human gene probe.
Off-target hits reflect human probes detecting a non-orthologous NHP gene, readily identified and filtered during standard analysis. Custom probe design is available for genes critical to your study that fall in the undetected fraction.
Rat tissue is not currently supported with a dedicated probeset. The rat genome is more divergent from mouse than NHP is from human, resulting in only ~63% predicted gene detection using the mouse probeset. A dedicated rat probeset from 10x Genomics is coming soon. Contact us to discuss your rat experiment requirements and timing.
VisiumHD + Single-cell RNAseq: a powerful combination
VisiumHD and single-cell RNAseq are complementary technologies. Spatial data maps where gene expression occurs across the tissue, while single-cell profiles resolve individual cell types and states within that context. Together they offer a more complete view of tissue architecture, cell types, cell states, and interactions than either platform alone.
Segmentation error introduces noise into spatial transcriptomic data — a substantial fraction of transcripts are detected within diffusion distance of cell boundaries, creating inherent ambiguity in cell-of-origin assignment that affects all current spatial transcriptomics platforms.