
Understanding tumor biology is critical for elucidating the mechanisms that drive cancer. The tumor microenvironment is a complex tissue containing a diversity of cell types that interact with each other to regulate cancer progression. The heterogeneity within this area makes it difficult to characterize using traditional approaches that result in a loss of resolution or spatial information.
Spatial organization of the tumor and its microenvironment has been linked to drug resistance and clinical outcomes in different types of cancer. Characterization of the spatial map of patient tumors could become a critical element for prognosis and treatment decisions. Using 10X Visium, researchers can profile the whole transcriptome of a tumor microenvironment while retaining high-resolution spatial information
Hunter et al (2021) used spatial transcriptomics in a zebrafish model of melanoma. Whole-animal tissue sections were processed with 10X Visium to study gene expression of the tumors as well as surrounding tissue.
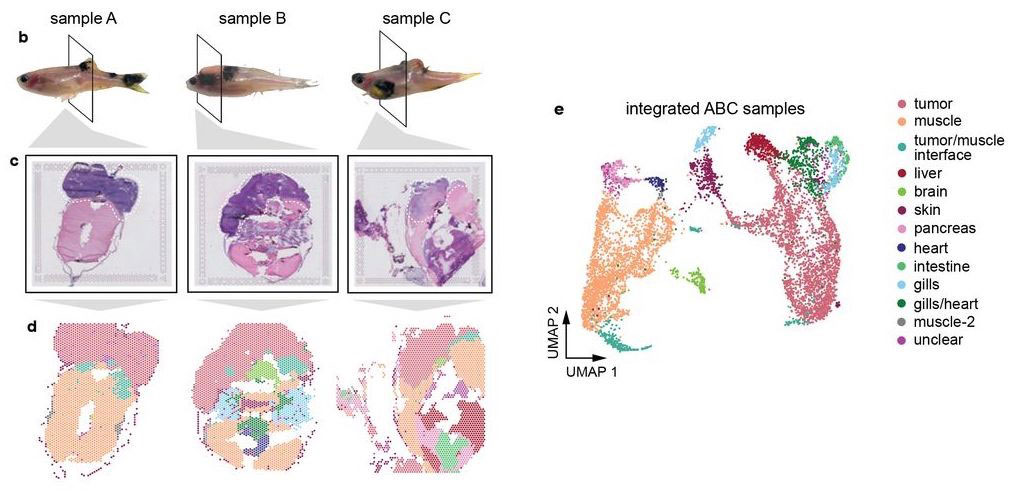
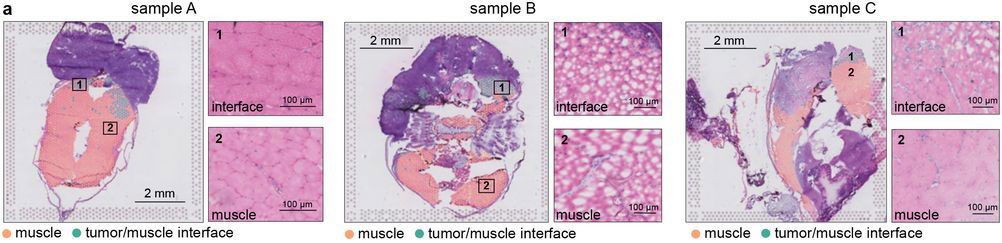
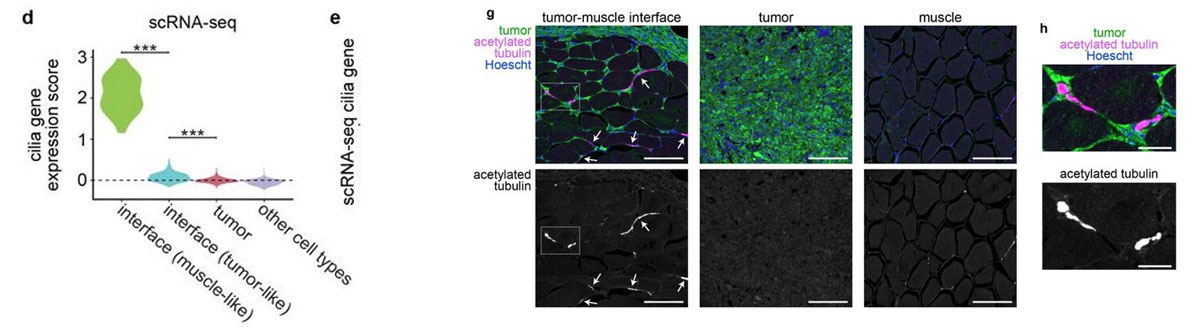
Most of the tissue was composed of two categories, one large group of tumor spots (pink) and another group of muscle spots (orange). Transcriptome data revealed a unique group of spots (teal) that cluster separately and are spatially located at the boundary between muscle and tumor. The authors termed this area the “interface” between muscle and tumor.
Although the “interface” spots are transcriptionally distinct, histological analysis reveals nothing visibly unique in the tissue in this area. Instead, these cells appear mostly indistinguishable from muscle cells.
Hunter et al (2021) also performed single-nuclei RNA-seq on these tissues in order to more deeply characterize the transcriptome. Looking at the “interface” area, the authors found an enrichment of cilia genes in the single-nuclei RNA-seq data. They also confirmed the presence of cilia proteins at the front of the tumor where it invades muscle tissue. Although the muscle and tumor themselves are not ciliated, this “interface” is, and the authors are now studying how cilia could be driving growth of tumors into the neighboring tissue.
Spatial information is critical in the study of neurodegenerative diseases, most of which still have few treatment options.
Protein co-detection by immunofluorescence can help the study of pathology of neurodegenerative illnesses. For instance, co-detection of Lewy bodies in patients with Parkinson’s disease or of amyloid plaques in Alzheimer’s disease gives their precise location within a tissue section. This allows researchers to characterize rare or novel cell types surrounding the area of pathology, further deepening our understanding of the mechanisms of these disorders. Spatial transcriptomics could also help identify potential new biomarkers which might aid in developing effective treatments.
Chen et al (2020) used spatial transcriptomics to identify two gene networks induced by amyloid plaques in both mouse and human brain tissue.
First, the authors used Spatial Transcriptomics (ST) (an earlier method upon which 10X Visium is based) in a mouse model of Alzheimer’s disease. They collected sequential coronal sections of the brain to allow for ST as well as immunostaining of 6E10 (amyloid beta), GFAP (astrocytes), NeuN (neurons), and DAPI (nuclei). Looking at the immediate area around amyloid plaques (~100um diameter), they discovered two gene co-expression networks driven by deposits of amyloid beta: 1) a plaque-induced gene network (largely contributed by microglia), and 2) an oligodendrocyte gene network composed of several myelination genes.
To confirm their findings, Chen et al (2020) used in situ sequencing (ISS) to visualize expression of the plaque-induced genes, which verified that these alterations do occur at the single-cell level.
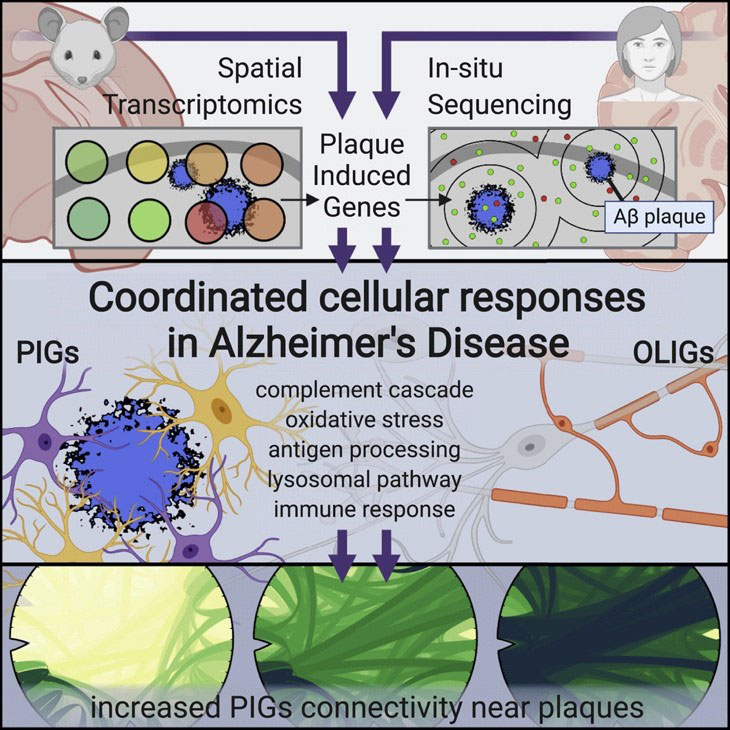
The authors also identified several plaque-induced genes in post-mortem human brain tissue from AD patients, suggesting these networks are conserved and likely relevant to human Alzheimer’s disease pathology.
Multiomic data integration gives you a more complete, multilayered analysis of your tissue. The tool we use at 3DG is Seurat, an R-based program with a number of tools for analyzing multi-modal single-cell data (https://satijalab.org/seurat/). Using Seurat, you can identify shared cell types or cell states between different datasets, across different conditions and technologies. In addition to spatial transcriptomics, you can also integrate single-cell RNA-seq (scRNA-seq) with other “omics” such as proteomic or chromatin accessibility data.
Visium spatial transcriptomic data often contain multiple cell types within the tissue area covered by a spot, resulting in a spot transcriptome that is a blend of multiple cells. Deconvoluting that spot transcriptome into its constituent cell type transcriptomes can be done using single-cell or single-nuclei transcriptome data. Beyond its improved resolution, another advantage of scRNA-seq data is significantly increased depth. On average, scRNA-seq experiments detect more genes per cell than spatial transcriptomics.
For this reason, when performing a spatial experiment, we recommend conducting a single-cell or single-nuclei experiment from the same tissue at the same time. This maximizes concordance between the two datasets, which helps aid integration. It also ensures that rare or unique cell populations from your tissue will be represented in the analysis, which may not be the case when using publicly available reference scRNA-seq datasets. If this is not an option for your tissue, there are several existing reference datasets that can be used for cell type annotation.
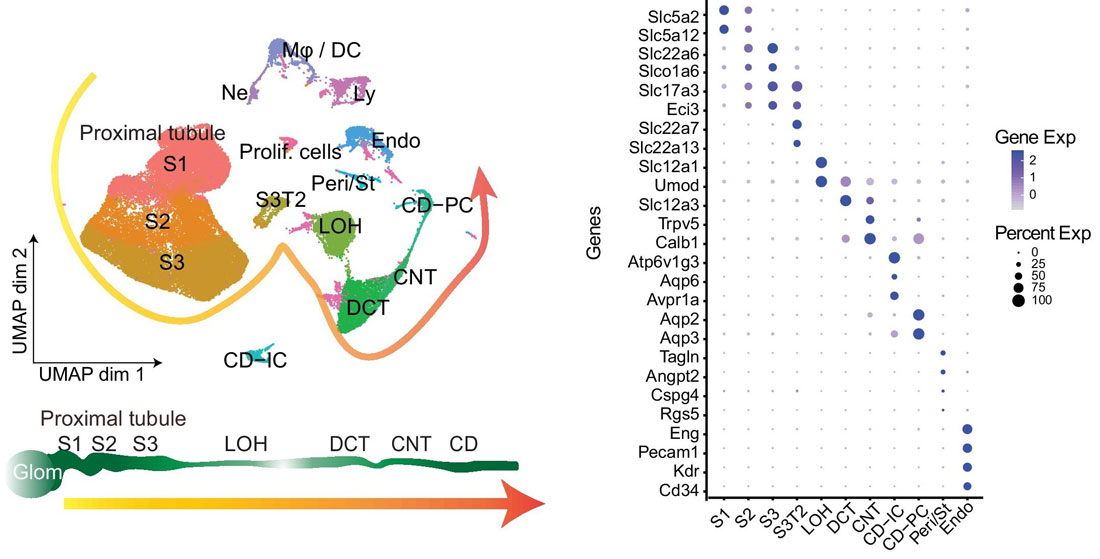
Before using your own single-cell RNA-seq dataset for integration, that data must be annotated. First, the cells are clustered and marker genes for each cluster are identified using Seurat’s FindMarkers() function. Cluster marker genes are then cross-referenced with known marker genes across various cell categories, and each cluster is annotated with a cell type. For example, in the figure shown below, ~63,000 mouse kidney cells were sequenced using 10X Genomics’ Chromium Single Cell 3’ Gene Expression Kit and annotated by identified marker genes. The left panel shows the UMAP of kidney single-cell clusters, while the right panel shows a dot plot of marker genes that characterize particular kidney cell types.
Once the scRNA-seq dataset is annotated, Visium spots can be deconvoluted using Seurat’s label transfer workflow. This method outputs a probabilistic cell type classification for each spot. The figure below shows a mouse kidney Visium spatial map before and after scRNA-seq integration.
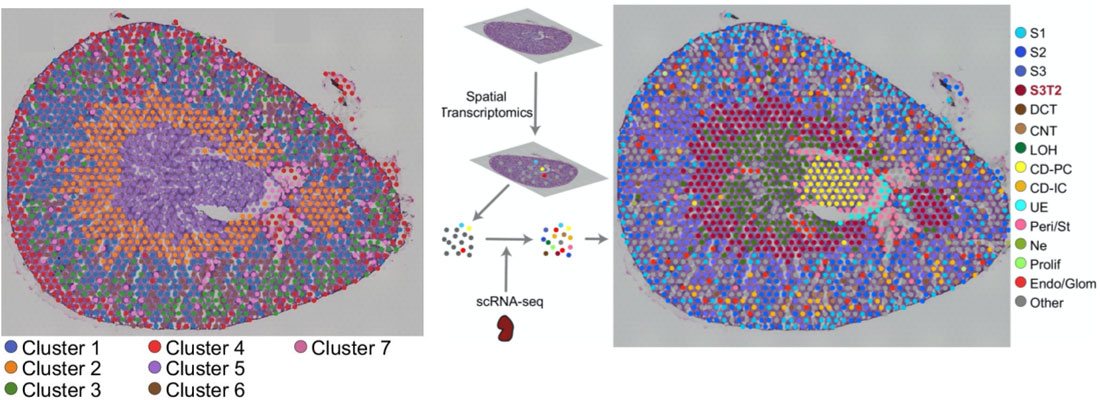
On the left, spots are color-coded by their unbiased clustering assignments. Using only Visium data, spots are separated into 7 gene expression groups (above, left panel). The authors then integrated their scRNA-seq data with the Visium data, and calculated prediction scores for cell types across all the Visium spots. Single cell RNA-seq integration improved the resolution of their kidney spatial map: instead of 7 clusters, the integrated dataset has 15 (above, right panel). Most notably, cluster 7 (left panel, purple) was clearly resolved into multiple cell types: Loop of Henle cells (right, green) and principal collecting duct cells (right, yellow).
An important consideration when integrating single-cell data is whether the datasets are matched (where multimodal data is recorded simultaneously from the same single cell) or unmatched (where data comes from different cells, but from the same sample, tissue, or organ). “Matched” technologies include CITE-seq (Cellular Indexing of Transcriptomes and Epitopes by Sequencing), scM&T-seq (single-cell Methylome and Transcriptome sequencing) and REAP-seq (RNA Expression And Protein sequencing), and more techniques are undoubtedly being developed.
Currently, most integrative analyses work with unmatched datasets due to technical limitations of matched single-cell experiments. However, analysis of matched data is particularly powerful due lower variation and batch effects.
Multidimensional molecular information is necessary to fully characterize cell states.
Immune cell subtypes, in particular, are often molecularly similar but functionally different. Single-cell technologies that go beyond the transcriptome are important for understanding such cell types. Furthermore, on a technical level it is often difficult to isolate immune cells such as T cells, due to low RNA quantity and high RNase levels.
To address these limitations, multimodal methods capture single-cell data from multiple types of molecules. For example, CITE-seq allows for simultaneous quantification of RNA and surface protein levels.
The latest version of Seurat introduced weighted nearest neighbor (WNN) analysis, a strategy for integrating multimodal data. WNN is an unsupervised method to assess information for each modality in each cell and then use cell-specific modality “weights” to allow for integrated analysis of multimodal data.
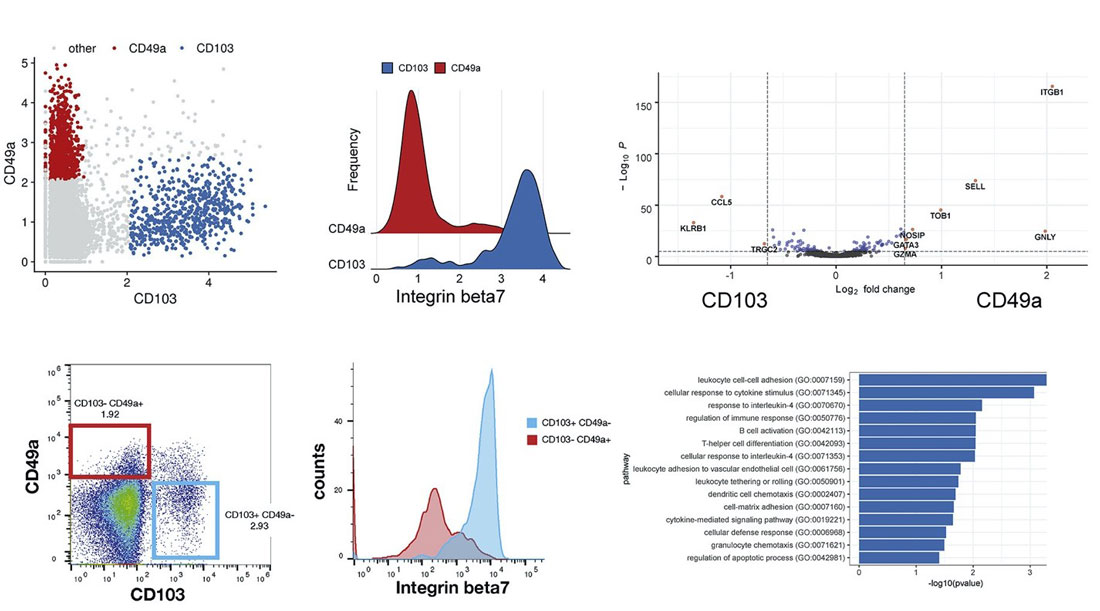
WNN was first described in Hao et al (2021), where the authors analyzed a CITE-seq dataset of ~200,000 human peripheral blood mononuclear cells (PBMCs) with 228 cell-surface protein markers. Using WNN, the authors were able to uncover levels of heterogeneity within lymphoid populations that were undetectable using scRNA-seq alone.
For example, CD8+ memory T cells clustered into multiple subpopulations driven by mutually exclusive expression of CD49a and CD103, both integrin proteins. Differential expression between CD49a+ and CD103+ CD8+ T cells revealed genes enriched for T cell activation, response to signals, and chemotaxis.
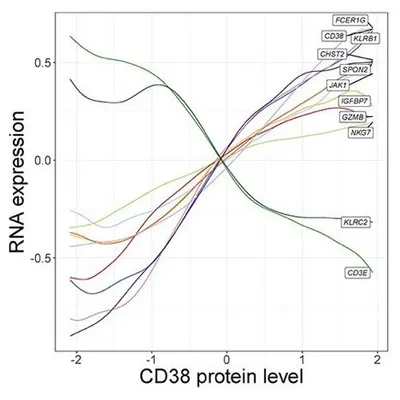
In addition, NK cells clustered into five groups that are driven by a two-dimensional gradient of CD16 and CD38 protein expression. The authors then looked at genes correlated with the CD38 protein gradient, which had never before been discovered. CD38 protein expression was positively correlated with expression of the signaling adaptor FCER1G, but negatively correlated with NK activating receptor gene KLRC2. Interestingly, this gene expression profile is similar to “adaptive” NK cell responses seen in individuals infected with cytomegalovirus (CMV). These results show the power of multimodal analysis for studying single-cell states.
As current single-cell technologies advance in scale, resolution, and the ability to acquire simultaneous molecular information, multimodal computational tools will be needed to derive the most power from these large-scale datasets. WNN is an integrative analysis method that can be used to analyze three or more simultaneously measured modalities across single-cell datasets. It is a powerful tool that is generally applicable to any high-dimensional single-cell datasets.